Welcome to Zijun Zhang's Homepage!

My name is Zijun Zhang (张子钧; also go by Frank). Currently I am a Flatiron Research fellow with Dr. Olga Troyanskaya at Princeton University/Simons Foundation. My research interests span machine learning, human genetics/transcriptomics, statistics and bioinformatics.
I obtained my Ph.D. in Bioinformatics with Dr. Yi Xing at UCLA, with research focus on elucidating the regulatory basis of human transcriptomes. While at UCLA, I also pursued a Master's degree in Statistics with Dr. Ying Nian Wu , studying automated machine learning and deep learning methods. During my undergrad at Zhejiang University, I worked with Dr. Ming Chen for three years to study the metabolic networks using computational and systems biology approaches.
Deep-learning powered Knowledge-base Democratizes Big Data
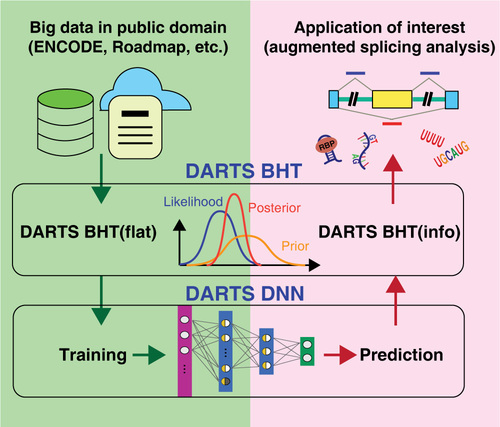
The rapid accumulation of RNA-seq data across diverse cell types and conditions provides an unprecedented resource for characterizing transcriptome complexity. However, the use of these large-scale data in routine RNA-seq studies to detect patterns of expression and thereby discover new regulatory events has been limited.
Conceptually, the DARTS framework transforms existing RNA-seq big data into a splicing knowledge-base by deep learning, and help democratize the information from large consortium projects to help individual investigators better characterize alternative splicing profiles using their specific RNA-seq datasets. Read More
Statistics Reveals Novel Regulatory Patterns
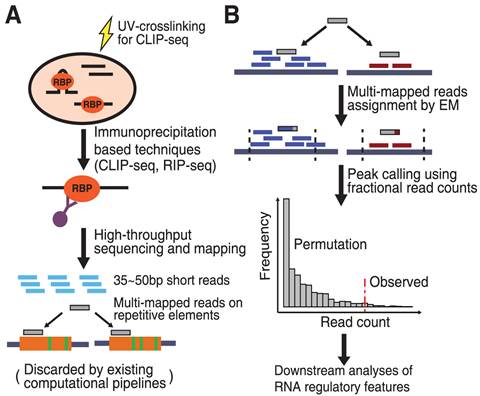
CLIP/RIP-seq allows transcriptome-wide discovery of RNA regulatory sites. As CLIP-seq/RIP-seq reads are short, existing computational tools focus on uniquely mapped reads, while reads mapped to multiple loci are discarded.
CLAM uses a statistical algorithm called "Expectation–Maximization" to assign multi-mapped reads and calls peaks combining uniquely and multi-mapped reads. CLAM provides a useful tool to discover novel protein–RNA interactions and RNA modification sites from CLIP-seq and RIP-seq data, and reveals the significant contribution of repetitive elements to the RNA regulatory landscape of the human transcriptome. Read More